Med DNA-sekvensering forstår vi bestemmelse av baserekkefølgen i et DNA-molekyl. Sangers kjedetermineringsmetode ligger til grunn for de fleste sekvenseringsteknikker som brukes i dag. Grunnlaget for metoden ligger i dideoksynukleotiders terminerende effekt på ulike DNA polymerasers aktivitet.
Templat (dobbelt- eller enkelttrådig DNA), primer, dNTP og en DNA polymerase uten exonuklease-aktivitet blandes med en liten andel av et dideoksynukleotid. Polymeriseringen vil terminere vilkårlig ved inkorporering av dideoksynukleotid istedet for det tilsvarende deoksynukleotid.
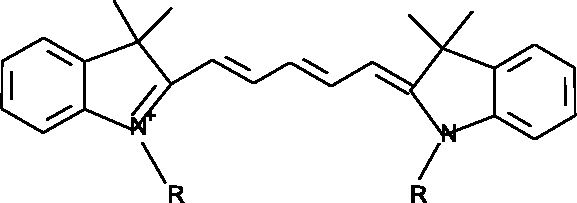
Separate reaksjoner for hvert av de fire nukleotidene vil gi distinkte blandinger av terminerte DNA-fragmenter som kan skilles ved elektroforetiske teknikker. Fragmentene kan detekteres ved ulike teknikker som radioaktiv eller fluoriserende merking. Bruk av fluoriserende primer eller dNTP gir mulighet for automatisering av sekvenseringen. En mye brukt fluoriserende kjemisk gruppe er Cy5, som er et indokarbocyanin (se figur 3.5)
Varmestabile polymeraser og PCR-teknologi har gjort sekvensering til en meget følsom teknikk med mindre strenge krav til mengde templat. Sekvenseringsreaksjonene kjøres i sykler av denaturering, annealing og elongering, som gir lineær amplifisering av terminerte fragmenter. Denne varianten av sekvensering kalles ofte ``cycle sequencing''
Prosedyren nedenfor er basert på protokollen for PERKIN
ELMER`S AmpliCycle kit (#N808-0175), og gjør på bruk av [ ![]() P]-dCTP eller -dATP.
P]-dCTP eller -dATP.
|
|
Nedenfor er innholdet i de kommersielle løsningene levert med ``Cycle sequencing kitet'' og gelløsningene gjengitt.
22,5 ![]() M
c7-dGTP
M
c7-dGTP
10 ![]() M
dATP, dCTP, dTTP
M
dATP, dCTP, dTTP
600 ![]() M
ddATP
M
ddATP
22,5 ![]() M
c7-dGTP
M
c7-dGTP
10 ![]() M
dATP, dCTP, dTTP
M
dATP, dCTP, dTTP
300 ![]() M
ddCTP
M
ddCTP
22,5 ![]() M
7-deaza-dGTP (c7-dGTP)
M
7-deaza-dGTP (c7-dGTP)
10 ![]() M
dATP, dCTP, dTTP
M
dATP, dCTP, dTTP
80 ![]() M
ddGTP
M
ddGTP
22,5 ![]() M
c7-dGTP
M
c7-dGTP
10 ![]() M
dATP, dCTP, dTTP
M
dATP, dCTP, dTTP
900 ![]() M
ddTTP
M
ddTTP
0,25 U/![]() l
AmpliTaq DNA polymerase CS
l
AmpliTaq DNA polymerase CS
500 mM Tris-HCl, pH 8,9
100 mM KCl
25 mM
MgCl2
0,25% (v/v) Tween 20
42,5 % Formamid
20 mM EDTA
0,05 % Bromfenolblått
0,02 % Xylene
Cyanol FF
5,7% (w/v) akrylamid
0,3% (w/v) methylen-bis-akrylamid
urea
(konsentrasjon ikke oppgitt, hemmeligholdt av produsenten)
0,1 M Tris-borat
- 2 mM EDTA buffer (pH 8,3)
Innholdet hemmeligholdes av produsenten.
Prosedyren som følger er basert på AMERSHAMs ``Thermo Sequenase fluorescent labelled primer cycle sequencing kit'' (RPN 2436) og protokoll for bruk av PHARMACIAs ALFExpress automatiske sekvenator.
Bruk Cy5-merket primer med lengde på minst 18 basepar. Primeren fortynnes til
brukskonsentrasjon på 1-2 ![]() M.
Templatet kan være enkelttrådig eller dobbeltrådig DNA av høy kvalitet. Primer
bør være komplementær til et område i templatet minst 40 basepar fra starten av
den ønskede sekvensen.
M.
Templatet kan være enkelttrådig eller dobbeltrådig DNA av høy kvalitet. Primer
bør være komplementær til et område i templatet minst 40 basepar fra starten av
den ønskede sekvensen.
|
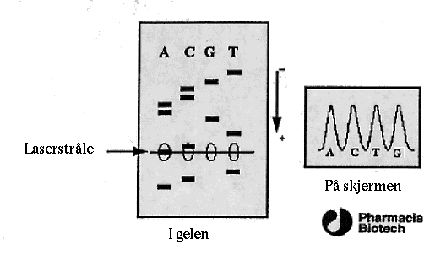
Nedenfor er fremgangsmåten ved støping av sekvensgel til ALFexpress automatisk sekvenator beskrevet. Både Long Ranger akrylamid og vanlig akrylamid (se punkt 3.3.1.3.5) kan benyttes til støping av gel. Med Long Ranger gel og godt templat kan man på det beste lese opp til 1000 basepar sekvens med metoden. Vanlig akrylamidgel gir bedre oppløsning enn Long Ranger gel i området 0-300 basepar
|
Nedenfor er montering av gel, igangsettelse av elektroforese og datalogging for ALFExpress beskrevet. Prosedyren gjelder ved bruk av programvare-pakken ALFWin for MS Windows 95. Prinsippet for ALFExpress er utdypet i figur 3.6.
Nedenfor er innholdet i de kommersielle løsningene levert med ``Thermo Sequenase fluorescent labelled primer cycle sequencing kit'' (RPN 2436) fra AMERSHAM gjengitt.
Tris-HCl (pH 9,5)
MgCl2
Tween 20
Nonidet P-40
2-mercaptoetanol
dATP, dCTP, dGTP, dTTP
ddATP
Termostabil pyrofosfatase
Thermo Sequenase DNA polymerase
Tris-HCl (pH 9,5), MgCl2, Tween 20, Nonidet P-40 , 2-mercaptoetanol, dATP, dCTP, dGTP, dTTP, ddCTP, Termostabil pyrofosfatase, Thermo Sequenase DNA polymerase
Tris-HCl (pH 9,5), MgCl2, Tween 20, Nonidet P-40 , 2-mercaptoetanol, dATP, dCTP, dGTP, dTTP, ddGTP, Termostabil pyrofosfatase, Thermo Sequenase DNA polymerase
Tris-HCl (pH 9,5), MgCl2, Tween 20, Nonidet P-40 , 2-mercaptoetanol, dATP, dCTP, dGTP, dTTP, ddTTP, Termostabil pyrofosfatase, Thermo Sequenase DNA polymerase
Formamid
EDTA
Metylfiolett
I det følgende er oppskriften på gelløsningene og bufferne anbefalt til ALFExpress gjengitt.
37,5 ml 40% Akrylamid
25 ml 10xTBE![]() (se
nedenfor)
(se
nedenfor)
105g urea
H2O til 250 ml
Lagres i kjøleskap på
lystett flaske
30 ml 50% Long Ranger Akrylamid
30 ml 10xTBE![]() (se nedenfor)
(se nedenfor)
105g urea
H2O til 250 ml
Lagres i kjøleskap på lystett flaske
Merk at denne oppskriften er forskjellig fra vanlig 10xTBE (beskrevet i
tillegg A).
121,14 g Tris base
51,32 g borsyre
3,72 g EDTA
Filtreres gjennom
0,45![]() m filter for å unngå utfelling
m filter for å unngå utfelling
10 ml 100% etanol
15 ![]() l
iseddik
l
iseddik
2 ![]() l
Bind-Silan
l
Bind-Silan
Dette avsnittet behandler bruk av dataprogrammer til analyse av DNA-sekvens. Da disse metodene er av en annen karakter enn de øvrige i oppgaven, har jeg valgt å inndele beskrivelsen etter et noe annet mønster. Hver programgruppe beskrives for seg, med en forenklet prinsipiell innledning og siden en kortfattet omtale av bruken.
Programmene FastA og blast søker i databaser etter sekvenser som er homologe til en oppgitt sekvens (query). FastA søker i DNA-databaser, mens blast også kan søke i protein-databaser.
FastA søker etter likhet mellom en sekvens (query) og en gruppe sekvenser (database-sekvenser) ved hjelp av metoden til PEARSON & LIPMAN (1988) [110]. Først lager programmet en ``ordliste'' som inneholder alle mulige ``ord'' i query-sekvensen. Med ord forstår vi alle nukleotidsekvenser med lengde n, som settes av bruker. Posisjonen(e) hvor hvert ord forekommer i query-sekvensen lagres i ordlisten.
Deretter søker programmet gjennom alle DNA-sekvensene i de ønskede databasene og sammenligner hver enkelt med query-sekvensen. Hver sammenligning utføres ved at det søkes etter likhet med ordene i ordlisten. Når likhet med et ord påvises, gis det en score-verdi til sekvensen det sammenlignes med (normal score-verdi er lik for alle ord).
Etter sammenligning summeres alle de enkelte score-verdiene for en sekvens sammen. Sekvensene som gav høyest score, evalueres videre med en score-matrise som tillater konserverte utbytninger og identitet i sekvenser kortere enn ordlengden (< n). Total score for hver sekvens her lagres som init1.
Deretter undersøker programmet om noen av områdene med likheter i samme sekvens ligger inntil hverandre. En ny parameter, initn beregnes som et mål på størrelsen av de identiske strekkene. Til sist sammenstilles (alignes) query-sekvensen med de beste sekvensene så langt ved hjelp av prosedyren til CHAO, PEARSON og MILLER (1992) [111]. Sistnevnte prosedyre beregner også en parameter opt.
Programmet presenterer resultatet for bruker først ved et dotplot som viser query-sekvensen nedover og antall treff (likhet med ord i ordlisten) som funksjon av denne. Deretter listes sekvensene med høyest initn-verdi i rekkefølge sammen med sine respektive init1, initn og opt-paramaterne. Til sist vises sammenstillinger (alignments) av query-sekvensene med de beste funnene.
Med søkeprogrammet blast (basic local alignment search tool) kan query-sekvensen være enten DNA eller peptid. Det samme gjelder for databasen. Blast kan også søke i peptid-databaser med DNA-query-sekvens og motsatt. Da translaterer programmet DNA-sekvensen til peptid i alle faser før homologi-søk.
Algoritmen til blast er basert på ALTSCHUL ET AL. (1990) [112] og opererer med et begrep som kalles MSP (Maximal-scoring Segment Pairs). Et segment-par (SP) er et strekk med likhet mellom query-sekvensen og sekvensen det sammenlignes med. Blast søker å utvide segmentparene helt til likheten tar slutt. Deretter beregnes en score-verdi for segmentparet. Alle segmentpar som er store nok til at de ikke forekommer tilfeldig med en relativ frekvens større enn en forventningsverdi (expectation value), listes etter score-verdi.
Blast bruker mye kortere tid på homologi-søk enn fastA, men er mindre sensitiv. FastA er bedre i stand til å detektere homologi på tross av innsetninger, utbytninger og strekk uten idenitet internt i homologe sekvenser.
FastA og blast er en del av gcg-pakken. Denne er tilgjengelig blant annet på serveren bioslave ved Bioteknologisenteret i Oslo. Alle kommandoer som gis i operativsystemet UNIX er her og i det følgende representert ved symbolet > og så selve kommandoen med annerledes font. Symbolet > representerer kommando-promptet og er ikke en del av kommadoen. For å oversende kommandoen til kommandotolker (shell) må return-tasten benyttes. Hjelp for alle kommandoene i gcg-pakken kan man få ved å gi kommandoen > genhelp [navn på kommando].
Compare er et program som brukes til å sammenligne to sekvenser med hensyn på homologi. Det søkes for homologi ved at et ``vindu'' på et visst antall baser (oppgis av bruker) flyttes langs begge sekvensene. Programmet kan finne punkter med identitet enten ved å detektere vinduer i begge sekvenser som har et antall like nukleotider over en viss grenseverdi eller ved å detektere helt identiske vinduer i begge sekvenser. Sistnevnte variant er 1000 ganger raskere enn førstnevnte. Merk at vinduet flyttes gjennom hele den ene sekvensen for hver vindu-posisjon i den andre sekvensen.
Resultatet av sammenligningen skrives til en fil som kan brukes til å lage et todimensjonalt plot med programmet dotplot. De to sekvensene plottes på hver sin akse. Forekomster av identitet mellom sekvensene vises ved et punkt i koordinatsystemet. For eksempel vil identitet mellom vinduet som starter i posisjon 20 i den ene x-sekvensen og i posisjon 350 i y-sekvensen, symboliseres ved punktet (20,350). Fullstendig identiske sekvenser resulterer i en rett linje diagonalt i koordinatsystemet.
Filer kan kopieres fra databaser og over til ditt filområde ved hjelp av kommandoen > fetch [filnavn]. Filene som skal sammenlignes bør ligge i samme katalog (i filsystemet), her bør man også være plassert ved start av programmene.
Programmet DNA Strider er et program for enkle analyser av DNA-sekvens med grafisk brukergrensesnitt. Visning av seter for restriksjonsenzymer, hypotetiske fordøyninger, lokalisering av åpne leserammer, translasjon og aminosyrer er noen av mulighetene som foreligger. Dette avsnittet berører bare de deler av programvaren som har vært brukt i min oppgave og er ikke tenkt å være noen slags form for fullstendig brukermanual.
DNA Strider finnes foreløpig kun kompilert for MacOS, men kan kjøres fra andre operativsystemer dersom Mac-emulatorer er installert. Programmet startes ved å klippe på ikonet for programmet.